DETERMINACIÓN DE METALES PESADOS EN
MATERIAL PARTICULADO ATMOSFÉRICO POR ESPECTROSCOPÍA DE ABSORCIÓN ATÓMICA:
VALIDACIÓN
María Fernanda Sarmiento Gamero 1,
Carlos Daniel Ramos Contreras 2, Sofía Lorena Flórez Pérez 3,
Francisco José Molina Pérez 4.
1Química,
Analista área fisicoquímicos laboratorio GAIA. maria.sarmiento@udea.edu.co
2Químico,
PhD Ing. Ambiental, Coordinador de área fisicoquímicos laboratorio GAIA. daniel.ramos@udea.edu.co
3Especialista
en Gestión Ambiental y Producción Más Limpia, Analista área fisicoquímico
laboratorio GAIA. sofialorena.florez@udea.edu.co
4Doctor
en Ingeniería, Director técnico del laboratorio GAIA. francisco.molina@udea.edu.co
Grupo de
Investigación en Gestión y Modelación Ambiental-GAIA,
Escuela Ambiental, Facultad de Ingeniería, Universidad de Antioquia UdeA, Calle
70 #52 51, Medellín, Colombia.
RESUMEN
El análisis de Cr, Cd, Pb y Ni en
material particulado (PM10) permite establecer los niveles de exposición y de
cuantificación, empleando digestión ácida asistida por microondas y
espectroscopía de absorción atómica. Se evaluaron los parámetros: linealidad,
límite de detección y de cuantificación, precisión y veracidad. Se obtuvo
comportamiento lineal (R2>0,995) entre 0,75-2,5 μgCd, 8,75-125 μgNi, 5-25
μgCr y 8,75-15 μgPb. También, se evidenció efecto matriz (α=0.05) en todos los
metales estudiados. Los coeficientes de variación (0,9-12%) y los porcentajes
de recuperación (66-126%) se encontraron dentro de los intervalos permitidos.
El análisis del material de referencia (NIST 1649b Urban Dust) permitió
determinar la veracidad de la metodología (71-102 % R). Por consiguiente, la
metodología puede ser implementada para evaluar de manera confiable, el
cumplimiento de la resolución 2254:2017 del Ministerio de Medio Ambiente y
Desarrollo Sostenible.
Palabras clave: Material
particulado, metales, absorción atómica.
Recibido: 10 de junio de 2021. Aceptado: 16 de
agosto de 2021
Received: June 10, 2021.
Accepted: August 16, 2021
DOI: https://doi.org/10.33571/rpolitec.v17n34a10
DETERMINATION OF HEAVY METALS IN
ATMOSPHERIC PARTICULATED MATERIAL BY ATOMIC ABSORPTION SPECTROSCOPY: VALIDATION
ABSTRACT
The analysis of Cr, Cd, Pb, and Ni in
particulate matter (PM10) makes it possible to establish the levels
of exposure and risk to the health of the population. This work presents the
validation and implementation of analytical methodologies, using microwave-assisted
acid digestion and atomic absorption spectroscopy. The parameters were
evaluated: linearity, the limit of detection and quantification, precision, and
veracity. Linear behavior (R2> 0.995) was obtained between 0.75-2.5 μg Cd,
8.75-125 μg Ni, 5-25 μg Cr and 8.75-15 μg Pb. Also, a matrix effect (α = 0.05)
was evidenced in all the metals studied. The coefficients of variation
(0.9-12%) and the recovery percentages (66-126%) were found within the allowed
intervals. The analysis of the reference material (NIST 1649b Urban Dust)
allowed to establish the veracity of the methodology (71-102 % R). Finally, the
methodology can be implemented to reliably assess compliance with resolution
2254: 2017 of the Ministry of the Environment and Sustainable Development.
Keywords: Particulate matter,
metals, atomic absorption.
Cómo citar este artículo: M.F. Sarmiento,
C.D. Ramos, S.L. Florez, F.F. Molina. “Determinación de metales pesados en
material particulado atmosférico por espectroscopía de absorción atómica:
validación” Revista Politécnica, vol.17, no.34 pp.153-169, 2021.
DOI: https://doi.org/10.33571/rpolitec.v17n34a10
1.
INTRODUCCIÓN
La contaminación del aire se define como la presencia
de cualquier gas o partícula que, a una concentración suficientemente alta,
puede ser perjudicial para la vida, el medio ambiente y/o la infraestructura.
Los contaminantes que llegan al aire pueden ser emitidos por fenómenos
naturales (volcanes, incendios forestales, descomposición de materia orgánica,
entre otros) y por actividades humanas (procesos de combustión, actividades
industriales y transporte) [1]. Las fuentes de contaminación atmosférica se dividen
en fuentes fijas, las cuales hacen referencia a emisiones de procesos
industriales, canteras, minería, calderas, entre otras y las fuentes móviles
hacen referencia principalmente a vehículos de todo tipo [2]. Las fuentes fijas y móviles producen contaminantes
del tipo primario, estos entran directamente a la atmósfera, como el monóxido
de carbono, los hidrocarburos, el material particulado, el dióxido de azufre y
los óxidos de nitrógeno, que se conservan tal como fueron emitidos, también hay
del tipo secundario que corresponden a los originados cuando los primarios
reaccionan en la atmósfera [3].
De los contaminantes mencionados anteriormente, el
material particulado es de especial interés debido a que es una mezcla de
partículas líquidas y sólidas, de sustancias orgánicas e inorgánicas; que están
en suspensión en el aire con una composición muy variada, donde se pueden
encontrar componentes como sulfatos, nitratos, amoníaco, entre otros; los
cuales se presentan en una mayor proporción, en relación con otros elementos
como plomo (Pb), cadmio (Cd), cromo (Cr), zinc (Zn) y magnesio (Mg) [4],
[5]. El material particulado se clasifica según su
diámetro aerodinámico como un indicador del tamaño de las partículas; entre más
pequeño sea su diámetro mayor será su capacidad de entrar al sistema
respiratorio[5],
[6].Por consiguiente, según el tamaño de las partículas se han clasificado en:
partículas suspendidas totales (PST), las cuales se encuentran en el aire sin
importar su tamaño; partículas inhalables en suspensión, estas son conocidas
como PM10 cuyo diámetro es menor o igual a 10 μm; partículas finas o
respirables se conocen como PM2.5 cuyo diámetro es menor o igual a
2,5 μm y partículas ultrafinas cuyo diámetro es menor o igual 0,1 μm [1].
Muchos estudios realizados en ciudades
industrializadas han demostrado que el material particulado contiene elementos
como C, S, Ni, Pb, Cd, Fe, Cr, As, y demás; los cuales son nocivos en
concentraciones elevadas porque pueden ser absorbidos y acumulados en los
sistemas biológicos, y de esta manera, incrementan su toxicidad [7]–[9]. La presencia de dichos elementos en el organismo
causan problemas a la salud como retraso del desarrollo metal e intelectual en
los niños; en el caso de los adultos, ocasionan hipertensión y enfermedades
cardiovasculares por la presencia de plomo, afección de las vías respiratorias,
aparición de úlceras y dermatitis por exposición a cromo; la exposición a
níquel puede generar cáncer de pulmón y problemas dérmicos; la presencia de
cadmio ocasiona problemas en el hígado y riñón puesto que son los órganos
donde se concentra [8],
[10]–[12].
Por lo anterior, se hace necesario el diseño de una
metodología que permita cuantificar las concentraciones de los metales en el
material particulado de una manera simple y precisa.
La espectroscopía de absorción atómica (AAS), es una
técnica utilizada para la determinación de metales presentes en diferentes
matrices, en la cual los átomos gaseosos libres absorben la radiación
electromagnética a una longitud específica y así generan una señal medible que
es proporcional a la concentración de cada metal[13],
[14]. Además de la AAS, existen diferentes técnicas para la
cuantificación de metales como: la emisión atómica (AES), espectroscopía de
fluorescencia (AFS), espectroscopía de absorción atómica con horno de grafito
(GFAAS), espectrometría de masas de plasma acoplada inductivamente (ICP-MS),
espectrometría de emisión óptica plasmática inductivamente acoplada (ICP-OES) y
fluorescencia de rayos X (XRF); sin embargo, estas presentan muchas
desventajas, entre ellas, altos costos que están asociados al uso de reactivos
e instrumentación necesaria para el análisis, poca precisión y altas
interferencias[15],
[16].
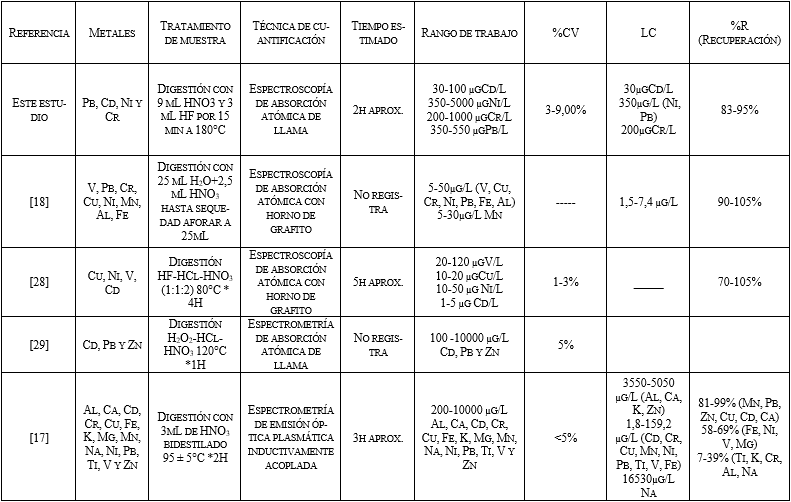
De las técnicas mencionadas para la cuantificación de
metales en material particulado PM10, algunas de las que se han
validadas son, la espectroscopía de emisión atómica de plasma acoplado
inductivamente (ICP-OES) [17]
y la espectroscopía de absorción atómica con horno de grafito [18],
con las que se realiza una comparación con la metodología validada en este
trabajo Tabla 5.
El presente trabajo tiene como objetivo validar una
metodología donde se utiliza parámetros como linealidad, efecto matriz, límite
de detección y cuantificación, precisión y veracidad; con el fin de establecer
si es aplicable al evaluar el cumplimiento de los niveles máximos permisibles
de exposición a estos metales definidos en la resolución No. 2254 del 01 de nov
de 2017.
2.
MATERIALES Y MÉTODO
Con el propósito de determinar la concentración de
Cr, Pb, Cd y Ni en los filtros de material particulado PM10 proporcionados
por el Área Metropolitana del Valle de Aburrá (AMVA), se valida una
metodología en la cual se utilizan secciones circulares (ϕ:1.9cm) de los
filtros de material particulado (PM10), los cuales son sometidos a
un digestión ácida asistida por microondas con el fin de descomponer totalmente
la matriz y así tener lo elementos de interés disponibles para realizar la
cuantificación por espectroscopía de absorción atómica de llama, empleando
lámparas de cátodo hueco específicas para cada elemento.
2.1. Muestreo
Las muestras de material particulado se recolectaron
empleando muestreadores de alto volumen Graseby-Andersen GBM2360 los cuales
eran programados para tomar muestras de aire a un flujo promedio de 1,13 m3/min
durante un periodo de 24 horas en filtros de cuarzo (203 x 254 mm, Whatman)
para un volumen total promedio de 1627,2 m3. Los filtros fueron precalentados a 400°C por 12 horas
para eliminar posible contaminación orgánica. Después de la recolección, las
muestras son nuevamente acondicionadas y almacenadas a -30°C para análisis
posteriores.
2.2. Materiales
y equipo
Las rectas de calibración se prepararon utilizando
soluciones patrón certificadas trazables a NIST de 1.000 mg/L de Cd, Ni, Pb y
Cr (Panreac, España), la digestión ácida asistida por microondas (ETHOS One®
Milestone, Italia.) de los filtros de material particulado se realizó siguiendo
la metodología propuesta por la Agencia de Protección Ambiental Americana (EPA)
[19],
en la cual se empleó ácido nítrico al 65% (EMSURE® Merck, Alemania) y ácido
fluorhídrico al 40% (Panreac, España). Y la cuantificación se realizó mediante un
espectrofotómetro de absorción atómica (iCe 3000 SERIES, Thermo Scientific,
USA). El procedimiento se muestra en la Fig. 1.
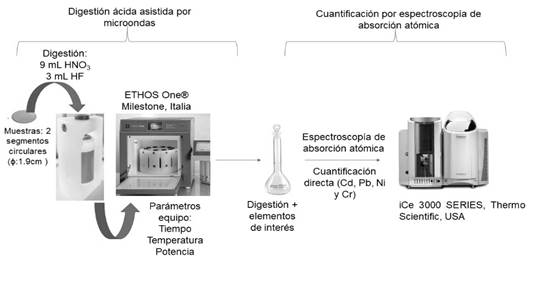
Fig. 1. Diagrama de análisis de metales en PM10
por absorción atómica.
2.3. Optimización
instrumental
Se realizó la optimización del espectrofotómetro de
absorción atómica, preparando las soluciones a las concentraciones sugeridas
por el software SOLAAR del espectrofotómetro iCE 3000 SERIES Thermo Scientific
y se realizaron modificaciones del flujo del gas y la velocidad de bombeo hasta
obtener la mejor respuesta.
2.4. Validación
de la metodología
Se evaluaron los parámetros de validación:
linealidad, límite de detección y cuantificación, precisión, veracidad, rango
de trabajo e incertidumbre.
2.4.1. Linealidad
Para los metales Cd, Cr, Ni y Pb se analizaron 6
niveles cuyas concentraciones estudiadas fueron las siguientes para Cd (0,75,
1,00, 1,25, 1,63, 2,00 y 2,50 μg), para Cr (5,00, 8,75, 12,50, 16,50, 21,25 y
25,00 μg), para Ni (8,75, 10,00, 12,50, 16,25, 21,25 y 25,00 μg), para Pb
(8,75, 10,00, 11.25, 12,50 y 13,75 μg). Se realizó la regresión lineal graficando la
concentración μg de cada elemento analizado vs.
absorbancia obtenida cuya expresión es descrita en la Ecuación 1.

Donde 𝑦
corresponde a la variable dependiente (absorbancia), y 𝑏 a la variable independiente
(concentración en μg del elemento), x la pendiente de la curva de regresión y 𝑎 el intercepto.
2.4.2. Efecto
matriz
Para determinar el efecto que aporta la matriz de
material particulado al resultado del análisis, se evaluó la linealidad a
través de la preparación de las curvas de calibración empleando muestras
típicas digeridas para aforar cada uno de los niveles estudiados para los
elementos de interés.
2.4.3. Límite
de cuantificación (LC) y detección (LD)
Para la determinación de los límites, se
analizaron 10 muestras de blanco y se calcularon a partir de la expresión de la
señal ruido (Ecuación 2)

Donde:
K: constante que usualmente se considera 3 para el
límite de detección y 10 para el límite de cuantificación.
s: desviación estándar de los blancos.
b: pendiente de la curva de calibración [20].
Los resultados obtenidos al aplicar la ecuación
corresponden a los límites teóricos. Se comprobó el límite de cuantificación
obtenido por análisis de quintuplicados, donde se prepararon estándares con
concentraciones cercanas al valor obtenido.
2.4.4. Precisión
Fue evaluada a dos niveles: repetibilidad y
precisión intermedia. La repetibilidad se realizó por quintuplicado, en
tres niveles (bajo, medio y alto) para lo cual se enriquecían dos secciones
circulares de las muestras de material particulado adicionando una
concentración conocida para posteriormente determinar el coeficiente de
variación (%CV) Ecuación 3.
Las concentraciones evaluadas fueron 0,75, 1,25,
2,50 μg para Cd; 5,00, 12,50 y 25,00 μg para Cr; 8,75, 12,50 y 25,00 μg para Ni
y 8,75, 11,25 y 13,25 μg para Pb.

Donde s es la desviación estándar de las medidas y
ẍ es el promedio de los datos. Para la precisión intermedia se repitió el mismo
procedimiento de preparación de muestras y de cuantificación, solo que en esta
ocasión el procedimiento fue realizado por otro analista. A partir de los resultados obtenidos por el
analista responsable de la validación y el segundo analista, se evaluó la
precisión intermedia del método mediante el cálculo del %CV global.
2.4.5. Veracidad
Se realizó el mismo procedimiento seguido para la
evaluación de la repetibilidad del método y se calculó el porcentaje de
recuperación en los niveles evaluados. Los porcentajes de recuperación se calculan
mediante la Ecuación 4.
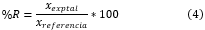
Donde 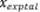 se refiere a la cantidad del
analito determinada experimentalmente y
se refiere a la cantidad del
analito determinada experimentalmente y  corresponde a la concentración
adicionada. Adicionalmente se evaluó la veracidad del método
empleando el material de referencia certificado trazable a NIST 1649b Urban
Dust. Se pesaron 0,30 g del material de referencia y los porcentajes de
recuperación se calcularon empleando la Ecuación 4, esto se realizó por
triplicado. Y como criterio de aceptación se utilizó la guía
ISO 33 (2015) [21],
la cual plantea la Ecuación 5 para establecer si el método es veraz o no. Se
emplea un factor de cobertura k=2.
corresponde a la concentración
adicionada. Adicionalmente se evaluó la veracidad del método
empleando el material de referencia certificado trazable a NIST 1649b Urban
Dust. Se pesaron 0,30 g del material de referencia y los porcentajes de
recuperación se calcularon empleando la Ecuación 4, esto se realizó por
triplicado. Y como criterio de aceptación se utilizó la guía
ISO 33 (2015) [21],
la cual plantea la Ecuación 5 para establecer si el método es veraz o no. Se
emplea un factor de cobertura k=2.
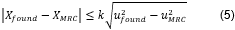
 = concentración hallada del MRC (mg/kg)
= concentración hallada del MRC (mg/kg)
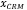 = concentración del MRC en el certificado
(mg/kg)
= concentración del MRC en el certificado
(mg/kg)
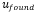 = incertidumbre hallada del MRC
= incertidumbre hallada del MRC
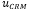 = incertidumbre del MRC en el certificado
= incertidumbre del MRC en el certificado
2.4.6. Incertidumbre
La incertidumbre del método se calculó a través de
las directrices establecidas por Eurachem [22].
De esta forma se determinaron las fuentes de incertidumbre que más aportan al
resultado final del mesurando, siguiendo la ley de propagación de errores (Ecuación
6 y Fig. 2). La incertidumbre combinada estimada se expande a
un intervalo de aproximadamente al 95% de confianza mediante el uso del factor
de cobertura k=2 (Ecuación 7).


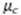 Corresponde a la incertidumbre combinada
donde
Corresponde a la incertidumbre combinada
donde 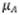 y
y 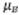 son las incertidumbres
individuales de cada una de las fuentes que tienen un aporte significativo,
son las incertidumbres
individuales de cada una de las fuentes que tienen un aporte significativo, 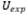 es
la incertidumbre expandida y k el factor de cobertura
es
la incertidumbre expandida y k el factor de cobertura
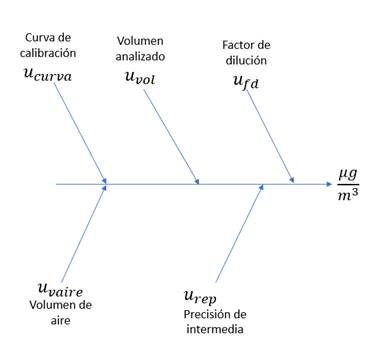
Fig. 2. Diagrama causa efecto para las principales
fuentes de incertidumbre consideradas en el análisis.
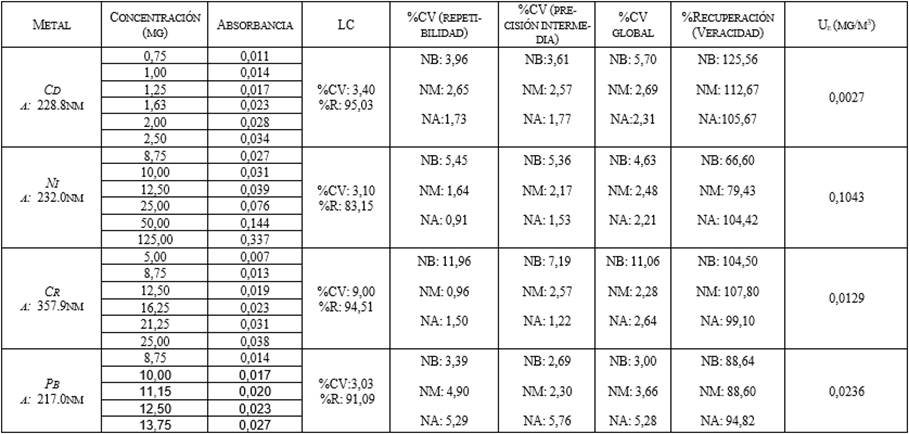
Tabla 1. Valores obtenidos para la linealidad,
repetibilidad, precisión intermedia, recuperación e incertidumbre para las
metodologías evaluadas. NB: nivel bajo, NM: nivel medio, NA: nivel alto.
3.
RESULTADOS
Al evaluar la relación existente entre la absorbancia
obtenida con respecto a cada una de las concentraciones estudiadas para los
distintos metales, se obtuvo un comportamiento lineal en todos los casos, esto indica
que, al no estar en presencia de la matriz, el instrumento arroja una respuesta
lineal a cambios en las concentraciones de los elementos y se obtiene un
coeficiente de determinación mayor o igual 0,995 en todos los casos.
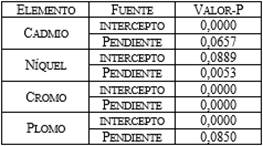
El análisis de varianza (Tabla 2) realizado para cada
uno de los metales estudiados indicó que la variabilidad de la absorbancia
puede ser explicada por el factor concentración (μg) (p< 0,05) por lo tanto,
existe una relación entre las dos variables con un nivel de confianza del 95,0%.
Tabla 1. Análisis comparativo (ANOVA) entre las
regresiones lineales para los elementos estudiadas (α=0,05).
Para la evaluación del efecto que tienen los
componentes de la matriz en el análisis los metales se valoró la linealidad
preparando los estándares de las curvas de calibración a partir de filtros de
material particulado digeridos. La construcción de la curva se realizó con las
soluciones madre preparadas para la evaluación de la linealidad.
Al realizar el análisis de regresión lineal para cada
uno, se encontró que todos los metales tienen efecto matriz dado que, al
realizar la comparación entre las pendientes de las curvas preparadas con agua
destilada y la curva preparada con material particulado digerido para cada uno
de los metales estudiados se evidenciaron diferencias significativas
(p<0,05), por lo tanto, se esperaría que en la rutina de análisis sea
necesario hacer la curva de calibración sobre la matriz y preparar un blanco de
matriz. Fig. 3 , 4, 5, y 6.
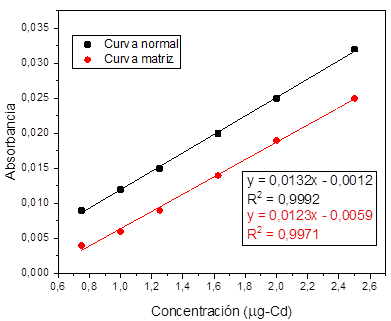
Fig. 3. Linealidad y efecto matriz para cadmio.
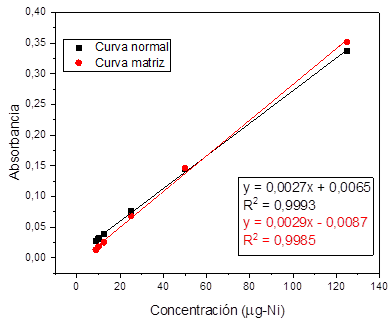
Fig. 4. Linealidad y efecto matriz para níquel.
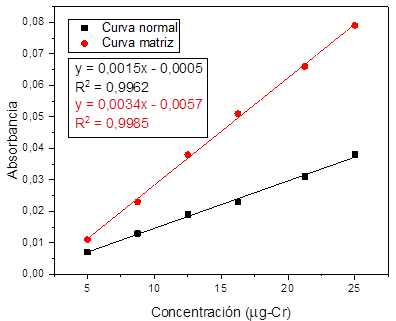
Fig. 5. Linealidad y efecto matriz para cromo.
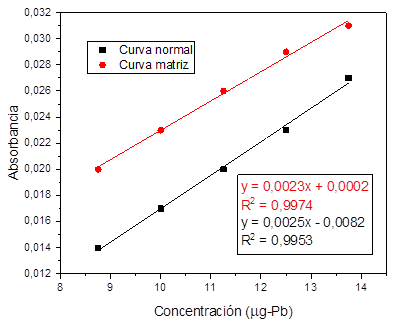
Fig. 6. Linealidad y efecto matriz para Pb.
Para la metodología desarrollada se pueden detectar
de forma exploratoria concentraciones desde 0,1568 μg de Cd, 0,2811 μg de Ni,
0,1333 μg de Cr y 1,563 μg de Pb. Posteriormente, se realizó la verificación de
los límites de cuantificación establecidos en la metodología, de los
coeficientes de variación y los porcentajes de recuperación reportados en la
Tabla 1. Al evaluar el grado de dispersión de los datos
(repetibilidad) con base al coeficiente de variación (CV) se obtuvieron valores
menores al 15% para repeticiones realizadas por un mismo analista a las mismas
condiciones, estos resultados se encuentran dentro de los intervalos aceptados [23].
La evaluación de la precisión intermedia, realizada
por un segundo analista, proporcionó coeficientes de variación menores al 15%
para los niveles de concentraciones evaluados, los resultados se muestran en la
Tabla 1, los cuales se encuentran entre del intervalo considerado como
aceptable (CV <15%) [23]. Al comparar con otra metodología como la
espectrometría de emisión atómica de plasma acoplado inductivamente (ICP-OES)
se encontraron resultados similares, puesto que, los coeficientes de variación
(%CV) oscilan entre 1,0-12,7 para los elementos estudiados [24].
Las fuentes de incertidumbre que se tuvieron en
cuenta para el cálculo de las contribuciones fueron: incertidumbre por
interpolación de la curva de calibración 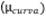 , volumen de muestra analizado
, volumen de muestra analizado 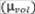 , factor
de dilución
, factor
de dilución 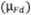 , volumen de aire
, volumen de aire  y reproducibilidad
del método según la Ecuación 8.
y reproducibilidad
del método según la Ecuación 8.
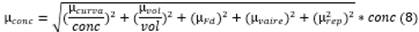
4.
DISCUSIÓN
El análisis de efecto matriz mostró interferencias
por parte de la matriz en los resultados obtenidos, esto puede estar
relacionado con la complejidad de la matriz que puede contener materia
orgánica, partículas que formen compuestos con los metales estudiados e
interfieran en su cuantificación. Además, la presencia de silicatos interfiere
en el análisis de cadmio, la presencia de hierro y níquel en la matriz
interfieren en la cuantificación de cromo y las concentraciones elevadas de
cromo y hierro interfieren en la cuantificación de níquel[25],
[26].
Al evaluar el límite de cuantificación se encontró
que, para valores inferiores de concentración de 0,75 μg de Cd, 8,75 μg de Ni,
5,00 μg de Cr y 8,75 μg de Pb se obtienen coeficientes de variación por fuera
de los criterios establecidos por la Association of Official Analytical
Chemists AOAC (<15%), por lo tanto, estos niveles fueron definidos como
límites de cuantificación. El análisis de repetibilidad del método evaluada como
coeficiente de variación (CV) permitió obtener valores entre los intervalos
esperados para las concentraciones evaluadas con base a los lineamientos de la
Association of Official Analytical Chemists (AOAC) [23].
Esto significa que la metodología diseñada presenta una precisión admisible a
nivel de repetibilidad.
Estudios realizados con técnicas como la
espectroscopía de emisión óptica con plasma acoplado inductivamente (ICP-OES
mostraron coeficientes de variación (0.60-2.97) comparables con los reportados
en este estudio [17].
El efecto del factor analista (u operario) en la variabilidad de los resultados
obtenidos para la metodología fue establecido (Tabla 1). Se encontraron
coeficientes de variación globales entre 2,00 y 11,00, los cuales cumplen
también con el criterio de un CVglobal< 2CV (repetibilidad), este
resultado permitió confirmar que el grado de dispersión de los resultados es
aceptable y por lo tanto, se puede decir que, el cambio de analista no
afectaría el desempeño del análisis[20].
Al evaluar la veracidad del método a través de
muestras enriquecidas con diferentes concentraciones que se encuentran en el
rango de trabajo (Tabla 1), se obtuvieron valores que se encuentran entre el
intervalo aceptable con base a lo establecido por la AOAC. Los porcentajes de
recuperación por encima del 100% pueden ser a raíz de la presencia de materia
orgánica u otras partículas que, ocasiona este incremento a pesar del análisis
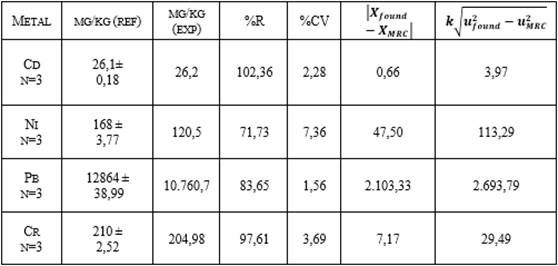
del blanco y de su sustracción a las concentraciones obtenidas [25]. Al analizar el material de referencia por triplicado
se encontraron los valores reportados en la Tabla 3, y al aplicar la ecuación 5
se obtuvo que todos los valores obtenidos cumplen con lo establecido en la Guía
ISO 33 (2015), por consiguiente, el método es veraz.
Tabla 3. Comparación de los valores experimentales
(exp) con los de referencia (ref) para NIST 1649b Urban Dust.
El análisis de las fuentes de incertidumbre
involucradas en el cálculo de la concentración se permitió evidenciar que el
mayor aporte está relacionado con la curva de calibración, debido a que, se
suman todas las contribuciones de la preparación de los puntos de cada nivel de
calibración (Fig. 7).
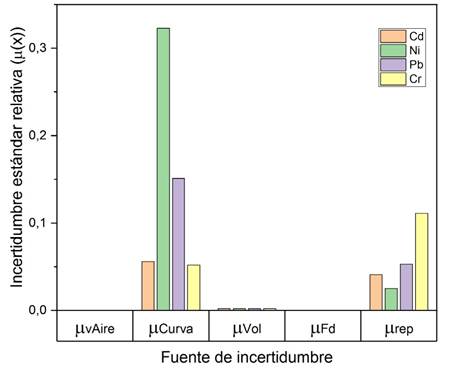
Fig. 7. Aportes de incertidumbre estimadas para cada
metodología.
Dado que los valores de los límites de cuantificación
son menores (al menos 10 veces) a los establecidos en la resolución No. 2254
del 01 de nov de 2017 para los metales de interés, se puede concluir que esta
metodología puede ser implementada para evaluar el cumplimiento de dicha
resolución (Tabla 4).
Tabla 4. Comparación con límites de cuantificación
con la normativa.
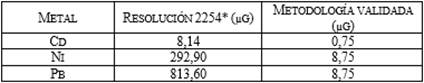
*Valores obtenidos teniendo en cuenta un volumen de
aire muestreado de 1627,2m3 de acuerdo con IDEAM [27].
Por último, al comparar la metodología validada con
otras metodologías se encontró que se emplean técnicas de cuantificación mucho
más sensibles con tratamientos de muestra que requieren mayor tiempo, generando
mayores costos, por tal razón, la metodología validada en este trabajo tiene un
beneficio económico considerable.
Con respecto a los porcentajes de recuperación, se
encuentra que son altos, lo que implica que la metodología es veraz al
compararlos con las otras técnicas, y a pesar de no ser una técnica tan
sensible como las demás se tienen límites de cuantificación y rangos de trabajo
que permiten evaluar el cumplimento de la resolución. Al comparar los coeficientes de variación se
encuentra que, estos son altos, con respecto a las otras metodologías, pero
como se mencionó anteriormente, cumple con los lineamientos de la AOAC, en
consecuencia, se puede decir; que se tiene una metodología sencilla, precisa y
económica. En la Tabla 5 se muestra la comparación de las metodologías.
5.
CONCLUSIONES
La metodología validada para la
cuantificación de Pb, Cd, Ni, Cr y As en filtros que se utilizan para el
monitoreo de material particulado PM10 en el Área metropolitana del
Valle de Aburrá, implementada en el Grupo de Investigación y Modelación
Ambiental- GAIA-, proporciona resultados confiables; es decir, precisos y
veraces con base a los criterios de aceptación establecidos por medio de la
determinación de los parámetros de validación; linealidad, rango de trabajo,
límites de cuantificación y detección, precisión (repetibilidad y precisión
intermedia) y veracidad.
Esta metodología, también puede ser
implementada para evaluar el cumplimento de la Resolución No. 2254 del 01 de
noviembre de 2017 del Ministerio de Medio Ambiente y Desarrollo Sostenible en
la cual se encuentran los niveles máximos de exposición para estos metales,
puesto que los límites de cuantificación obtenidos son inferiores a lo
reportados en la norma.
Tabla 5. Tabla comparativa de metodologías
6.
AGRADECIMIENTOS
Los autores agradecen a Universidad de Antioquia y a
MinCiencias por el soporte financiero al proyecto N° 111574454999-CTO 770/2016:
“Estimación de riesgo de cáncer por exposición a reconocidos carcinógenos
asociados al material particulado respirable en el Área Metropolitana del Valle
de Aburrá (AMVA) Antioquia”.
7.
REFERENCIAS BIBLIOGRÁFICAS
[1] S. L. C. González, “Material particulado en el aire y su
correlación con la función pulmonar en personas que realizan actividad física
en la cicloruta en la localidad kennedy en bogotá: estudio descriptivo
transversal,” Universidad Nacional de Colombia, 2018.
[2] E. M. López et al.,
“Contaminación atmosférica y efectos sobre la salud de la población Medellín y
su área metropolitana,” Medellín. Colombia, 2007.
[3] Annenberg Learner, “Unit 11
- Atmospheric Pollution,” The Habitable Planet, pp. 1–35, 2017,
[Online]. Available:
http://www.learner.org/courses/envsci/unit/text.php?unit=11&secNum=10.
[4] Fundación para la Salud
Geoambiental, “Material Particulado.” https://www.saludgeoambiental.org/material-particulado
(accessed Jun. 05, 2019).
[5] INECC, “Qué son, cómo son y
cómo se originan las partículas.,” no. x, pp. 13–32, 2016.
[6] A. Legarreta, A. Corral, M.
Delgado, J. Torres, and J. P. Flores, “Material particulado y metales pesados
en aire en ciudades mexicanas,” CULCyT, vol. 56, no. 1, pp. 234–245,
2015, [Online]. Available:
http://erevistas.uacj.mx/ojs/index.php/culcyt/article/view/818.
[7] L. H. H. Luisa Fernanda
Perez Fadul, “Determinación de metales pesados en particulas respirables e
identificación de fuentes de emisión, a partir de un muestreo atmosférico en la
localidad de puente aranda en la ciudad de bogotá,” Universidad de la Salle,
2006.
[8] Y. C. Reyes, I. Vergara, O.
E. Torres, M. Díaz, and E. E. González, “Contaminación Por Metales Pesados:
Implicaciones En Salud, Ambiente Y Seguridad Alimentaria,” Ing. Investig. y
Desarro., vol. 16, no. 2, pp. 66–77, 2016, doi:
10.19053/1900771x.v16.n2.2016.5447.
[9] A. A. Gharaibeh, A. W. O.
El-Rjoob, and M. K. Harb, “Determination of selected heavy metals in air
samples from the northern part of Jordan,” Environ. Monit. Assess., vol.
160, no. 1–4, pp. 425–429, 2010, doi: 10.1007/s10661-008-0706-7.
[10] K. N. and A. Lanphear, B.P.,
Dietrich and P.C, “Cognitive deficits associated with blood lead concentrations
<10 microg/dL in US children and adolescents,” pp. 521–529, 2002.
[11] L. F. Londoño Franco, P. T.
Londoño Muñoz, and F. G. Muñoz Garcia, “Los Riesgos De Los Metales Pesados En
La Salud Humana Y Animal,” Biotecnoloía en el Sect. Agropecu. y
Agroindustrial, vol. 14, no. 2, p. 145, 2016, doi:
10.18684/BSAA(14)145-153.
[12] “Agency for Toxic Substances
& Desease Registry,” 2005.
https://www.atsdr.cdc.gov/phs/phs.asp?id=243&tid=44 (accessed Jun. 10,
2019).
[13] B. Fernández, L. Lobo, and
R. Pereiro, Atomic Absorption Spectrometry: Fundamentals, Instrumentation
and Capabilities, 3rd ed., no. 1. Elsevier Inc., 2018.
[14] J. H. and S. R. C. Douglas
A. Skoog, F, Principios de análisis instrumental, Sexta edic. 2007.
[15] A. B. M. Helaluddin, R. S.
Khalid, M. Alaama, and S. A. Abbas, “Main analytical techniques used for
elemental analysis in various matrices,” Trop. J. Pharm. Res., vol. 15,
no. 2, pp. 427–434, 2016, doi: 10.4314/tjpr.v15i2.29.
[16] K. Aftab et al., “Determination
of different trace and essential element in lemon grass samples by X-ray
fluorescence spectroscopy technique,” Int. Food Res. J., vol. 18, no. 1,
pp. 265–270, 2011.
[17] L. M. B. Ventura, B. S.
Amaral, K. B. Wanderley, J. M. Godoy, and A. Gioda, “Validation method to
determine metals in atmospheric particulate matter by inductively coupled
plasma optical emission spectrometry,” J. Braz. Chem. Soc., vol. 25, no.
9, pp. 1571–1582, 2014, doi: 10.5935/0103-5053.20140142.
[18] J. Herrera M. and S. Rodríguez
R., “Validación de un método de análisis para la determinación de metales
pesados en partículas PM10 colectadas en aire ambiente,” Tecnol. en Marcha,
vol. 23, no. 3, pp. 33–46, 2010, [Online]. Available:
http://revistas.tec.ac.cr/index.php/tec_marcha/article/view/71.
[19] P. Coelho and R. Desti,
“Microwave assisted acid digestion of siliceous and organically based
matrices,” no. December, pp. 1–20, 1996.
[20] AEFI: Asociación Española de
Farmaceúticos de la Industria, Validación de Métodos Analíticos. Barcelona,
2001.
[21] J. R. Sieber, “How to use
and how not to use certified reference materials in industrial chemical
metrology laboratories,” Powder Diffr., vol. 35, no. 2, pp. 104–111,
2020, doi: 10.1017/S0885715620000202.
[22] S. Rasul, A. M. Kajal, and
A. Khan, “Quantifying Uncertainty in Analytical Measurements,” J. Bangladesh
Acad. Sci., vol. 41, no. 2, pp. 145–163, 2018, doi:
10.3329/jbas.v41i2.35494.
[23] S. Method, P. Requirements,
and A. Nutritionals, “Appendix F: Guidelines for Standard Method Performance
Requirements,” 2016.
[24] T. Byrd, M. Stack, and A.
Furey, “An Analytical Application for the Determination of Metals in PM10,” Adv.
Air Pollut., 2011, doi: 10.5772/21301.
[25] Instituto de Hidrología
Meteorología y Estudios Ambientales IDEAM, “Determinación de metales pesados
totales con digestión ácida y solubles lectura directa por Espectrofotómetría
de absorción atómica,” p. 16, 2004, [Online]. Available:
http://www.ideam.gov.co/documents/14691/38155/Metales+en+agua+por+Absorción+Atómica..pdf/e233a63d-378c-4f83-9311-d9375043cf2a.
[26] D. M. Gómez H., “Validación
de la metodología por el método estándar 3111a – absorción atómica
para el análisis de metales pesados en muestras de aguas y aguas residuales,”
p. 60, 2011.
[27] Ministerio de Ambiente
Vivienda y Desarrollo Territorial, “Manual de operación de sistemas de
vigilancia de la calidad del aire,” p. 287, 2008, [Online]. Available:
https://www.minambiente.gov.co/images/AsuntosambientalesySectorialyUrbana/pdf/contaminacion_atmosferica/Protocolo_Calidad_del_Aire_-_Manual_Operación.pdf.
[28] N. C. and D. L. Alberto
Fernández C, Raiza Fernández, “Metals Determination in Atmospheric Particulates
by Atomic Absorption Spectrometry with Slurry Sample Introduction,” At.
Spectrom., vol. 12, no. 4, 1991, doi: 10.1039/a800834e.
[29] M. A. Awan, S. H. Ahmed, M.
R. Aslam, and I. A. Qazi, “Determination of Total Suspended Particulate Matter
and Heavy Metals in Ambient Air of Four Cities of Pakistan,” Iran. J. Energy,
vol. 2, no. 2, pp. 128–132, 2011.